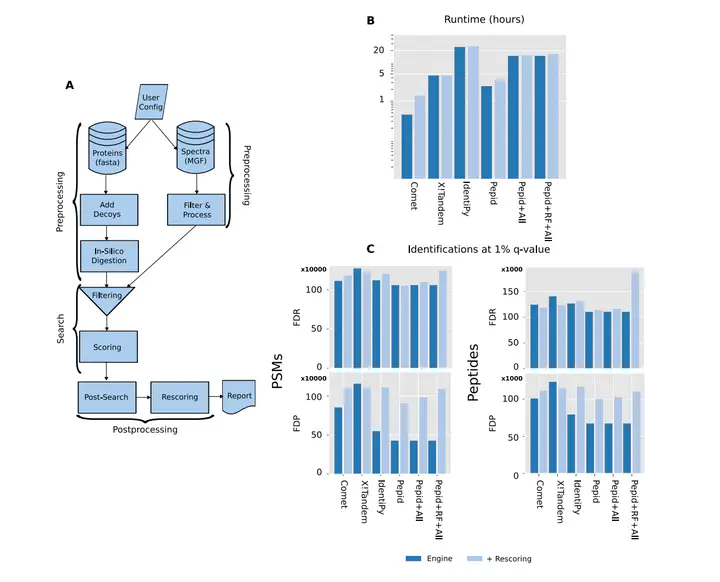
 Comparison between Pepid and commonly-used search engines on ProteomeTools first pool (1 458 831 spectra).
Comparison between Pepid and commonly-used search engines on ProteomeTools first pool (1 458 831 spectra).
Résumé
Current peptide search engines are optimized for wet-lab workflows, i.e. they operate in an “end-to-end” manner to achieve good identification results, not to be modified or provide algorithmic insight. This makes developing new software methods to solve problems in peptide identification methods difficult, often requiring a full engine rewrite. Recently, many deep learning methods were proposed as solutions to various parts of the peptide identification task, but virtually none of those methods have been implemented in any actual peptide search process. We believe that the lack of a reliable bioinformatics research platform for peptide identification that enables such integrations is slowing down proteomics research as a whole. We present pepid, a bioinformatics research-oriented peptide search engine. Unlike other search engines, pepid is specifically designed with ease of computational research in mind. Our design is highly flexible and allows easy modifications with little required software development expertise, allowing researchers to focus on analysing and improving peptide identification methods.It also takes recent computational trends into account, such as the recent slew of deep learning publications in proteomics, and features a multi-phased batched operations design that is more appropriate than the spectrum batch “end-to-end” designs of existing search engines for those approaches. We show that pepid is competitive with common engines in terms of both identification rates and runtime, forming a minimum required baseline to enable further identification research.
Jérémie Zumer
Étudiant au doctorat en informatique
Étudiant au Doctorat en Informatique | Amélioration de la spectrométrie de masse peptidique par l’apprentissage profond
Sébastien Lemieux
Chercheur principal
Chercheur principal, Unité de recherche en bio-informatique fonctionnelle et structurale, IRIC | Direction scientifique de la plateforme de Bio-informatique | Professeur agrégé, Département de biochimie et médecine moléculaire, Université de Montréal