Oral Presentation by Tom MacDougall at IUPAC | CCCE2021
Bravo to Tom MacDougall who has been accepted by IUPAC | CCCE2021 to give an oral presentation about Pairwise compound comparisons using machine learning makes more accurate and explainable property predictions (August 17, 2021, 11:30 AM – 12:20 PM ET)
Abstract
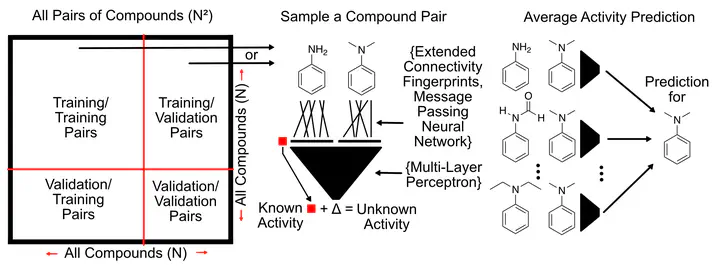
Structure-Activity Relationship (SAR) investigations are a multi-parameter optimization problem that can be overwhelming to all but the most experienced of chemists. Molecular Matched Pair Analysis (MMPA) tackles this problem, through the comparison of highly similar compounds, differing in structure by modification at one site. Through comparison and contrasting two compounds, more insight into the SAR landscape can be obtained. This work introduces the Pairwise Compound Comparison approach, which is based on MMPA but is generalized to any pair of compounds within a dataset.
The approach consists of a machine learning model that takes a pair of compounds as input instead of a single compound. In the training phase, pairs are sampled from the set of training compounds, and the model learns the activity difference between the two compounds. Through this mechanism, representations of pairs of compounds and the structural changes between them are learned and activity differences are predicted from these structural differences. The pairwise comparison approach can be invariant to the representation of the individual compounds, allowing the use of a wide range of different molecular representations. The compared representations in this work are two implementations of Message Passing Neural Networks (Neural fingerprints from Duvenaud et al. 2015 and Chemprop fingerprints from Yang et al. 2019), extended-connectivity fingerprints (ECFP), and a novel “collision-free” form of ECFP. “Collision-free” ECFP, as the name suggests are impervious to fingerprint hash collisions that affect regular ECFP. The developed pairwise models were validated using 5-fold cross-validation under two different data splitting scenarios, random splitting, as is typical of machine learning models, and scaffold splitting, which more closely resembles real-world scenarios.
Extensive hyperparameter optimization of the methods reveals vastly different optimum hyperparameters and model architectures when comparing the pairwise and single approaches. The larger input dimensionality of two compounds instead of one requires far more parameters to generalize well, and requires more training steps to optimize. The larger pair dataset of N2 pairs for N compounds results in far more training data points to train the larger models. The datasets used span different tasks in molecular property prediction including protein inhibition datasets from ChEMBL and MoleculeNet as well as Organic Photovoltaic properties from The Harvard Clean Energy Project.
The pairwise approach consistently achieves better R2 performance than the comparable single compound approaches on the 3 datasets when using ECFP as the individual compound representation, as well as when using collision-free ECFP. When using the fully differentiable message passing representations, the pairwise approach performs comparably to the single approach. In addition, the introduced collision-free fingerprints on their own are able to outperform the standard ECFP fingerprint while remaining more explainable due to the unambiguous substructure to fingerprint bit relationships. The performance improvement of the pairwise compound comparisons show the benefit of incorporating intuitive and proven SAR analysis approaches like MMPA into machine learning models.
Sébastien Lemieux
Principal Investigator
Principal Investigator, Functional and Structural Bioinformatics Research Unit, IRIC | Scientific direction of the Bioinformatics platform | Associate Professor, Department of Biochemistry and Molecular Medicine, Université de Montréal