Improving Peptide-Centric Mass Spectrometry with Deep Learning
Mass spectrometry is the most promising tool for proteomics research. However, state-of-the-art mass spectrometry analysis software leaves a lot to be desired. The two major peptide-centric mass spectrometry-based identification paradigms are de novo sequencing methods, which attempt to identify a peptide purely from its spectrum, and database search methods which require well-curated databases to work. Databases that are non-specific will result in too many false positives or false negatives. Those that are too big will cause current algorithms to mismatch incorrect sequences, and those that are too small will fail to obtain matches at a suitable false discovery rate cut-off. In addition, database-driven methods do not work properly when no database is available, as it is the case for MHC-I associated peptides.
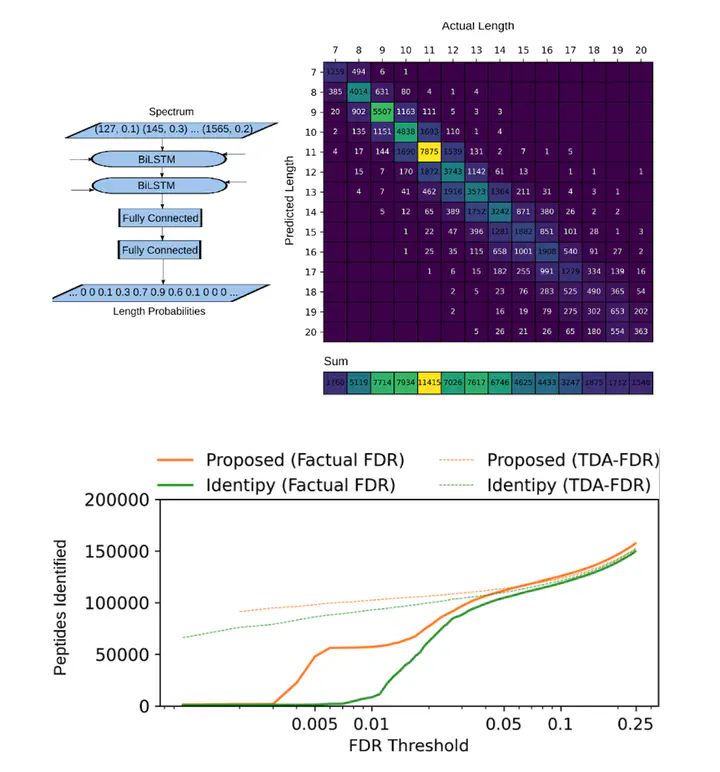
In Jérémie Zumer’s project, deep learning is used to bridge the gap by extracting never-before-available features from the mass spectra in a de novo manner. This is then used in a classical database-driven pipeline with an adjusted cost function that takes into account the added features. The result is greatly improved peptide identification rates on datasets such as ProteomeTools: almost 700% at a false discovery rate threshold of 1% compared to the IdentiPy software .